Introduction
- Amino acids are the building blocks of proteins and are essential for various metabolic processes.
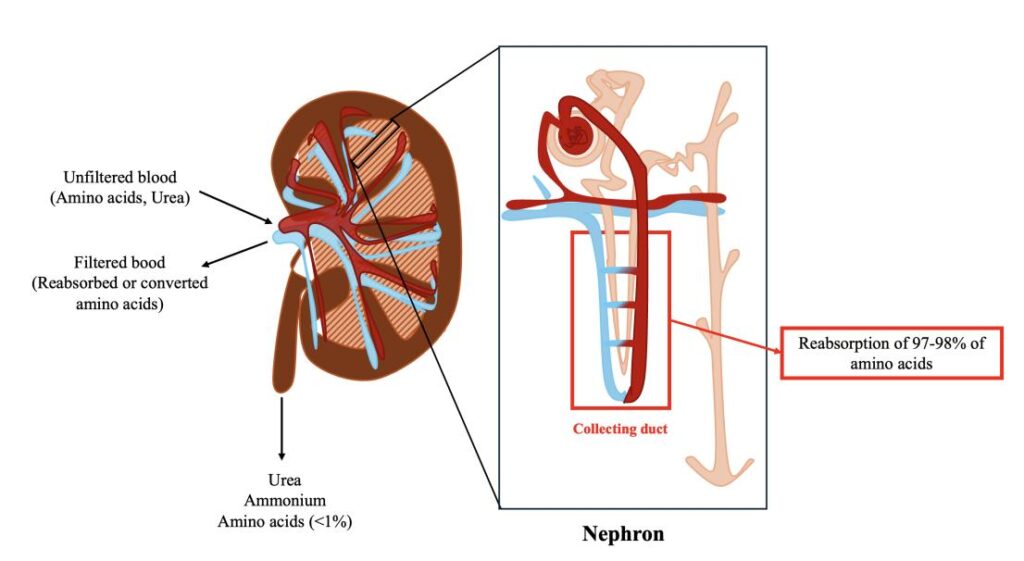
- Normally, amino acids are freely filtered by the glomeruli and almost completely reabsorbed in the proximal renal tubules.
- Therefore, only trace amounts of amino acids are present in normal urine.
- Aminoaciduria refers to the excessive excretion of one or more amino acids in urine.
- It is a biochemical finding rather than a disease itself.
- Aminoaciduria may result from:
- Inherited metabolic disorders
- Renal tubular transport defects
- Liver diseases
- Systemic disorders
- Toxic or drug-induced kidney damage
- Detection of aminoaciduria is important for diagnosing:
- Inborn errors of metabolism
- Renal tubular disorders
- Certain liver and systemic diseases
Historical Background
The clinical significance of aminoaciduria was first recognized in the early twentieth century with the discovery of inherited metabolic disorders such as cystinuria and phenylketonuria.
Major milestones include:
- 1810: Discovery of cystine urinary stones.
- 1908: Garrod described inborn errors of metabolism.
- 1934: Identification of phenylketonuria.
- 1950s–1960s: Development of amino acid chromatography.
- 1990s: Introduction of tandem mass spectrometry.
- Present era: Genetic sequencing and newborn screening programs.
These advances transformed aminoaciduria from a biochemical curiosity into an important diagnostic marker.
Physiology of Amino Acid
Understanding aminoaciduria requires knowledge of normal renal amino acid transport.
Glomerular Filtration
Most amino acids have low molecular weights and are freely filtered through the glomerular membrane.
Approximately:
- 500–700 mmol of amino acids are filtered daily.
- Nearly all filtered amino acids are reabsorbed.
Tubular Reabsorption
Reabsorption occurs primarily in the proximal convoluted tubule.
The process involves:
- Sodium-dependent transport systems
- Sodium-independent transporters
- Specialized carrier proteins
Reabsorption efficiency exceeds 98–99%.
Amino Acid Transport Systems
Neutral Amino Acid Transport System
Transports:
- Alanine
- Valine
- Leucine
- Isoleucine
- Tryptophan
- Phenylalanine
Defect leads to:
- Hartnup disease
Basic Amino Acid Transport System
Transports:
- Lysine
- Arginine
- Ornithine
- Cystine
Defect leads to:
- Cystinuria
Acidic Amino Acid Transport System
Transports:
- Aspartate
- Glutamate
Imino Acid Transport System
Transports:
- Proline
- Hydroxyproline
- Glycine
Defect leads to:
- Iminoglycinuria
Aminoaciduria
Aminoaciduria is defined as:
Excessive urinary excretion of one or more amino acids resulting from metabolic abnormalities, renal tubular transport defects, or both.
Classification
Aminoaciduria can be classified into several categories.
1. Overflow Aminoaciduria
Occurs when plasma amino acid concentration exceeds renal reabsorptive capacity.
Mechanism
- Increased amino acid production
- Defective amino acid metabolism
- Excessive dietary intake (rare)
The kidney functions normally, but excessive amino acids overwhelm reabsorption mechanisms.
Examples
- Phenylketonuria
- Maple Syrup Urine Disease
- Tyrosinemia
- Homocystinuria
- Hyperglycinemia
Characteristics
| Feature | Overflow Aminoaciduria |
|---|---|
| Plasma amino acids | Increased |
| Renal function | Normal |
| Tubular defect | Absent |
| Urinary amino acids | Increased |
2. Renal Aminoaciduria
Caused by defective tubular reabsorption.
Mechanism
Transport proteins are abnormal or damaged.
Characteristics
| Feature | Renal Aminoaciduria |
|---|---|
| Plasma amino acids | Normal |
| Renal transport | Defective |
| Tubular dysfunction | Present |
| Urinary amino acids | Increased |
3. Generalized Aminoaciduria
Involves multiple amino acids.
Usually associated with:
- Fanconi syndrome
- Heavy metal toxicity
- Wilson disease
- Drug-induced nephropathy
4. Selective Aminoaciduria
Only specific amino acids are excreted.
Examples:
- Cystinuria
- Hartnup disease
- Iminoglycinuria
Causes of Aminoaciduria
A. Genetic Causes
Phenylketonuria (PKU)
Deficiency of:
- Phenylalanine hydroxylase
Accumulation of:
- Phenylalanine
- Phenylpyruvate
- Phenylacetate
Results in aminoaciduria.
Maple Syrup Urine Disease (MSUD)
Deficiency of:
- Branched-chain α-ketoacid dehydrogenase complex
Accumulation of:
- Leucine
- Isoleucine
- Valine
Produces characteristic sweet-smelling urine.
Homocystinuria
Deficiency of:
- Cystathionine β-synthase
Accumulation of:
- Homocysteine
- Methionine
Tyrosinemia
Defects in tyrosine degradation enzymes.
Causes:
- Liver failure
- Renal dysfunction
- Aminoaciduria
Non-Ketotic Hyperglycinemia
Defect in glycine cleavage system.
Results in elevated urinary glycine.
B. Renal Tubular Disorders Causing Aminoaciduria
1 – Cystinuria
Genetic Basis
Mutations in:
- SLC3A1
- SLC7A9 genes
Amino Acids Affected
COLA amino acids:
- Cystine
- Ornithine
- Lysine
- Arginine
Clinical Features
- Recurrent kidney stones
- Hematuria
- Urinary obstruction
- Renal colic
Characteristic Finding
Hexagonal cystine crystals.
2 – Hartnup Disease
Genetic Defect
Mutation in:
- SLC6A19 transporter
Affected Amino Acids
Neutral amino acids.
Most important:
- Tryptophan
Consequences
Reduced niacin synthesis.
Clinical Features
- Pellagra-like rash
- Ataxia
- Psychiatric symptoms
- Photosensitivity
3 – Iminoglycinuria
Affects transport of:
- Glycine
- Proline
- Hydroxyproline
Usually asymptomatic.
4 – Fanconi Syndrome
Generalized proximal tubular dysfunction.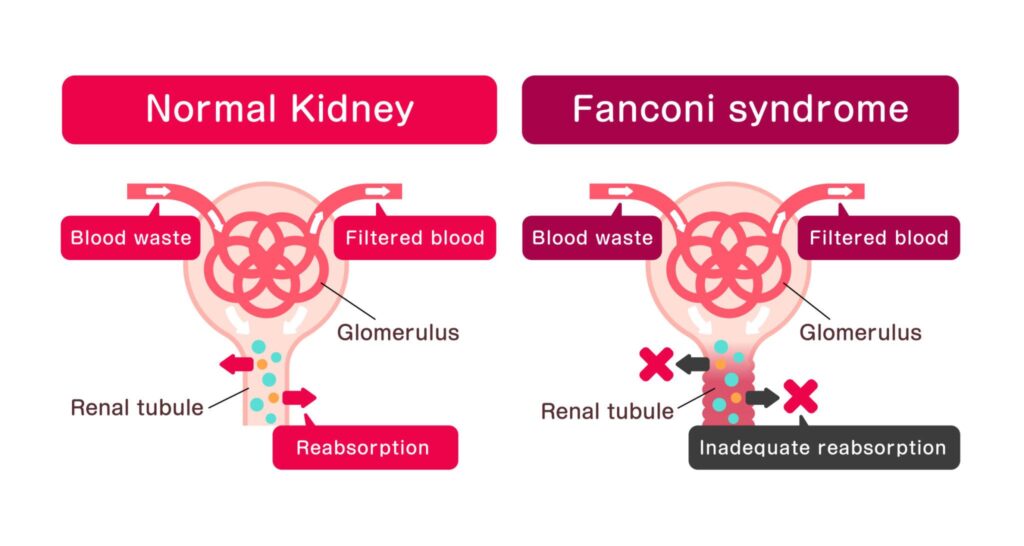
Causes
Inherited:
- Cystinosis
- Wilson disease
Acquired:
- Heavy metals
- Drugs
- Multiple myeloma
Associated Findings
- Aminoaciduria
- Glycosuria
- Phosphaturia
- Bicarbonaturia
- Uricosuria
Secondary Causes of Aminoaciduria
Liver Disease
Liver dysfunction alters amino acid metabolism leading to overflow aminoaciduria.
Examples:
- Cirrhosis
- Acute liver failure
Wilson Disease
Copper accumulation damages:
- Liver
- Kidneys
Produces generalized aminoaciduria.
Heavy Metal Poisoning
Includes:
- Lead
- Cadmium
- Mercury
Causes proximal tubular injury.
Drug-Induced Aminoaciduria
Associated drugs include:
- Ifosfamide
- Cisplatin
- Tenofovir
- Aminoglycosides
Clinical Manifestations
Symptoms depend upon the underlying disorder.
Renal Manifestations
- Kidney stones
- Hematuria
- Polyuria
- Proteinuria
Neurological Manifestations
- Intellectual disability
- Seizures
- Developmental delay
- Ataxia
Skeletal Manifestations
- Osteomalacia
- Rickets
- Growth retardation
Dermatological Manifestations
- Photosensitive dermatitis
- Pellagra-like lesions
Gastrointestinal Manifestations
- Vomiting
- Feeding difficulties
- Failure to thrive
Laboratory Diagnosis
1. Routine Urinalysis
- Initial screening test.
- Detects abnormal urinary constituents and crystals.
- May reveal:
- Cystine crystals (hexagonal crystals in cystinuria)
- Proteinuria
- Glycosuria (in Fanconi syndrome)
2. Qualitative Screening Tests
Cyanide-Nitroprusside Test
- Screening test for cystinuria and homocystinuria.
- Positive result produces a purple-red color.
3. Urine Amino Acid Analysis
- Confirms the presence of abnormal amino acid excretion.
- Identifies specific amino acids present in excess.
- Helps differentiate various aminoacidurias.
4. Chromatographic Methods
Paper Chromatography
- Traditional method for separating amino acids.
- Useful for preliminary identification.
Thin Layer Chromatography (TLC)
- Improved separation and visualization of amino acids.
High-Performance Liquid Chromatography (HPLC)
- Highly sensitive and accurate.
- Widely used for quantitative amino acid analysis.
5. Tandem Mass Spectrometry (LC-MS/MS)
- Modern gold-standard technique.
- Simultaneously detects multiple amino acids and metabolic disorders.
- Commonly used in newborn screening programs.
6. Plasma Amino Acid Analysis
- Measures amino acid concentrations in blood.
- Helps distinguish:
- Overflow aminoaciduria: Elevated plasma amino acids.
- Renal aminoaciduria: Normal plasma amino acids.
7. Renal Function Tests
- Blood urea nitrogen (BUN)
- Serum creatinine
- Estimated glomerular filtration rate (eGFR)
- Assess associated renal dysfunction.
8. Genetic Testing
- Identifies mutations causing inherited aminoacidurias.
- Useful for:
- Confirmatory diagnosis
- Carrier detection
- Prenatal diagnosis
Differential Diagnosis
Conditions producing increased amino acid excretion include:
- Cystinuria
- Hartnup disease
- Fanconi syndrome
- PKU
- MSUD
- Homocystinuria
- Tyrosinemia
- Wilson disease
- Cystinosis
- Heavy metal nephropathy
Treatment and Management
Treatment depends on the underlying cause of aminoaciduria.
1. Dietary Management
- Restriction of specific amino acids that accumulate in metabolic disorders.
- Specialized diets may be required.
Examples:
- Low-phenylalanine diet in Phenylketonuria (PKU)
- Restricted branched-chain amino acids in Maple Syrup Urine Disease (MSUD)
- Low-methionine diet in Homocystinuria
2. Vitamin Supplementation
- Certain disorders respond to vitamin therapy.
Examples:
- Pyridoxine (Vitamin B6) in Homocystinuria
- Niacin (Vitamin B3) in Hartnup Disease
3. Adequate Hydration
- Increased fluid intake helps dilute urine and reduce stone formation.
- Particularly important in Cystinuria.
4. Urinary Alkalinization
- Increases solubility of cystine and reduces stone formation.
- Commonly used agents:
- Potassium citrate
- Sodium bicarbonate
5. Specific Drug Therapy
- Used in selected metabolic disorders.
Examples:
- Nitisinone for Tyrosinemia Type I
- Cysteamine for Cystinosis
- Thiol-containing drugs (e.g., penicillamine) in severe Cystinuria
6. Treatment of Underlying Renal or Systemic Disease
- Management of Fanconi syndrome
- Treatment of Wilson disease
- Removal of nephrotoxic drugs
- Management of liver disease or kidney dysfunction
7. Genetic Counseling
- Recommended for inherited aminoacidurias.
- Helps with:
- Family screening
- Carrier detection
- Prenatal diagnosis
- Future pregnancy planning
Recent Advances in Aminoaciduria Research
1. Tandem Mass Spectrometry (LC-MS/MS)
- Highly sensitive and specific technique for amino acid analysis.
- Enables simultaneous detection of multiple metabolic disorders.
- Widely used in newborn screening programs.
2. Next-Generation Sequencing (NGS)
- Identifies mutations in genes responsible for amino acid transport and metabolism.
- Facilitates early and accurate diagnosis of inherited aminoacidurias.
- Supports personalized treatment strategies.
3. Metabolomics
- Comprehensive analysis of metabolites in biological samples.
- Provides detailed amino acid profiling.
- Helps identify novel biomarkers for diagnosis and disease monitoring.
4. Expanded Newborn Screening
- Early detection of disorders such as:
- Phenylketonuria (PKU)
- Maple Syrup Urine Disease (MSUD)
- Homocystinuria
- Tyrosinemia
- Allows prompt treatment and improved outcomes.
5. Precision Medicine
- Treatment tailored according to the patient’s genetic and metabolic profile.
- Improves therapeutic effectiveness and reduces complications.
6. Gene Therapy
- Emerging approach for correcting genetic defects causing inherited aminoacidurias.
- Currently under investigation for several metabolic disorders.
- Offers potential for long-term disease correction.
7. Improved Biomarker Discovery
- Identification of novel urinary and plasma biomarkers.
- Enhances early diagnosis, disease monitoring, and prognosis assessment.