Introduction
- Haemoglobin is the most important oxygen-carrying protein present in red blood cells (RBCs).
- It is responsible for transporting oxygen from lungs to tissues and carrying carbon dioxide from tissues back to lungs.
- Haemoglobin gives red blood cells their red colour.
- It is essential for normal respiration, cellular metabolism, and survival.
- Since every cell requires oxygen for energy production, haemoglobin plays a direct role in maintaining life.
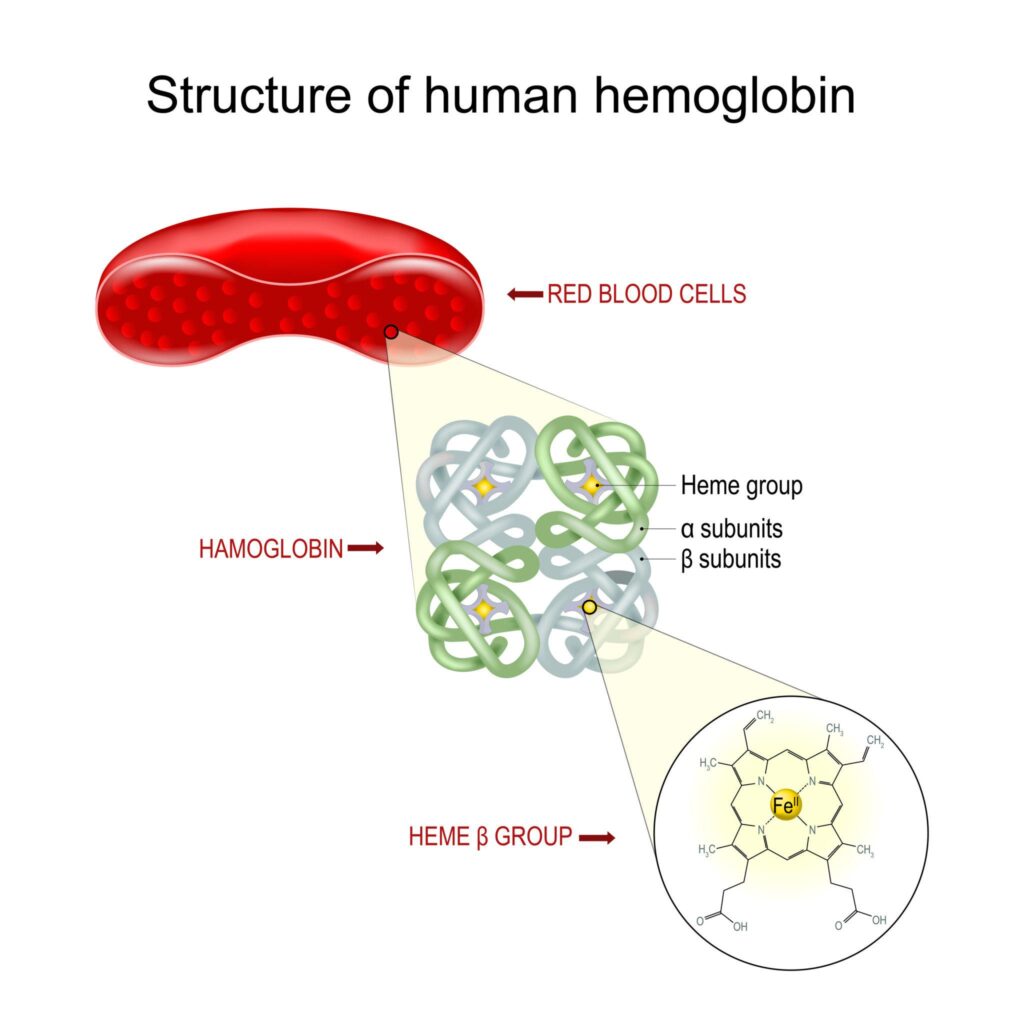
- Haemoglobin is a conjugated chromoprotein present inside erythrocytes.
- It is called conjugated protein because it contains:
- Protein part = Globin
- Non-protein prosthetic group = Heme
- Thus:
Haemoglobin = Globin + Heme
- It is a tetrameric protein because one haemoglobin molecule contains four globin chains.
Normal Haemoglobin Concentration
Normal haemoglobin concentration in blood:
| Group | Normal Hb Level |
|---|---|
| Adult Male | 14–16 g/dL |
| Adult Female | 13–15 g/dL |
| Newborn | 16–20 g/dL |
Low haemoglobin causes anaemia, while high haemoglobin may occur in dehydration or polycythaemia.
Structure of Haemoglobin
- Haemoglobin is a conjugated protein present in red blood cells (RBCs).
- It is made up of two main parts:
- Globin (protein part)
- Heme (non-protein prosthetic group)
Basic Composition
- One haemoglobin molecule contains four globin chains and four heme groups.
- Therefore haemoglobin is called a tetrameric protein.
- Molecular weight of haemoglobin is approximately 64,450 Dalton.
Globin Part
- The common adult haemoglobin is HbA.
- It consists of:
HbA=α2β2
- This means:
- 2 alpha (α) chains
- 2 beta (β) chains
Amino Acid Composition
- Each alpha chain contains 141 amino acids
- Each beta chain contains 146 amino acids
Total Amino Acids
- Total amino acids in one haemoglobin molecule = 574
Bonds Holding Chains Together
The four globin chains are held together by:
- Hydrogen bonds
- Ionic bonds
- Hydrophobic interactions
Heme Part
- Each globin chain contains one heme group.
- Therefore one haemoglobin molecule contains 4 heme groups.
Structure of Heme

- Heme consists of protoporphyrin IX ring with a central ferrous iron (Fe²⁺) atom.
- The porphyrin ring is made up of four pyrrole rings joined together.
Side Chains of Porphyrin Ring
- 4 methyl groups
- 2 vinyl groups
- 2 propionate groups
Iron Binding in Heme
- Iron forms six coordination bonds:
- Four bonds with nitrogen atoms of porphyrin ring
- One bond with globin histidine residue
- One bond with oxygen molecule
Oxygen Binding
- Each heme binds one oxygen molecule.
- Therefore one haemoglobin molecule binds four oxygen molecules.
1 Hb molecule=4 O2 molecules
Structural Feature
- Haemoglobin changes its shape during oxygen binding:
- T form (tense form) = deoxygenated form
- R form (relaxed form) = oxygenated form
Heme synthesis
- Heme synthesis is the stepwise formation of heme from simple precursor molecules.
- It occurs mainly in bone marrow and liver.
- The pathway takes place partly in mitochondria and partly in cytoplasm.
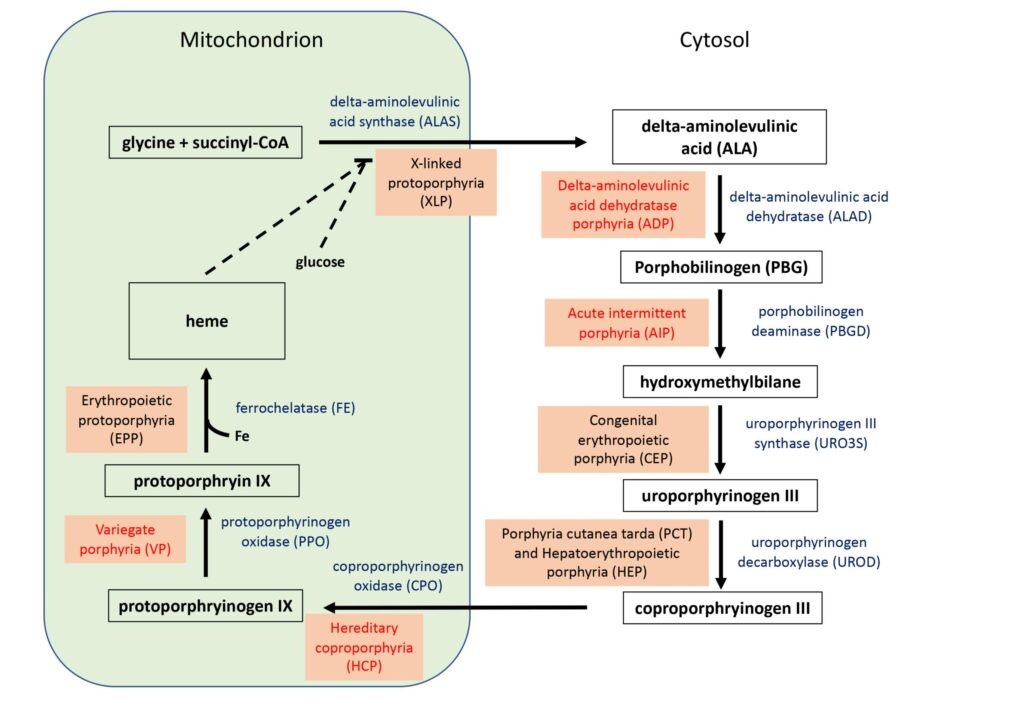
Stage 1: Formation of δ-Aminolevulinic Acid (ALA)
Glycine + Succinyl-CoA → δ-Aminolevulinic acid
- Glycine combines with succinyl-CoA.
- Enzyme: ALA synthase
- Cofactor required: Vitamin B6 (Pyridoxal phosphate)
- Site: Mitochondria
- This is the rate-limiting step of heme synthesis.
Stage 2: Formation of Porphobilinogen (PBG)
2 δ-ALA→Porphobilinogen
- Two molecules of ALA condense to form porphobilinogen.
- Enzyme: ALA dehydratase
- Site: Cytoplasm
Stage 3: Formation of Hydroxymethylbilane
- Four molecules of porphobilinogen combine to form a linear tetrapyrrole compound.
- Enzyme: Porphobilinogen deaminase
Stage 4: Formation of Uroporphyrinogen III
- Hydroxymethylbilane cyclizes to form uroporphyrinogen III.
- Enzyme: Uroporphyrinogen III synthase
Stage 5: Formation of Coproporphyrinogen III
- Uroporphyrinogen III undergoes decarboxylation.
- Enzyme: Uroporphyrinogen decarboxylase
Stage 6: Formation of Protoporphyrin IX
- Coproporphyrinogen III enters mitochondria again.
- It is converted into protoporphyrin IX through oxidation reactions.
Stage 7: Formation of Heme
Protoporphyrin IX+Fe2+→Heme
- Ferrous iron (Fe²⁺) is inserted into protoporphyrin IX.
- Enzyme: Ferrochelatase
- Site: Mitochondria
Regulation of Heme Synthesis
- Regulation of heme synthesis mainly occurs at the first step of the pathway.
- The most important regulatory enzyme is ALA synthase (δ-aminolevulinate synthase).
- This enzyme controls the rate of heme production.
Rate-Limiting Step
Glycine + Succinyl-CoA → ALA synthaseδ – ALA
- Glycine combines with succinyl-CoA to form δ-aminolevulinic acid (ALA).
- This is the rate-limiting step of heme synthesis.
- It occurs in mitochondria.
Feedback Inhibition by Heme
- Heme itself regulates its own synthesis.
- When heme concentration increases, it inhibits ALA synthase.
Effect
- ALA synthase activity decreases
- Heme synthesis slows down
Importance
- Prevents excess heme production
Regulation in Liver
- In liver, heme synthesis adjusts according to requirement of cytochromes.
- Certain drugs increase demand for cytochrome enzymes.
Drugs that stimulate heme synthesis
- Barbiturates
- Anticonvulsants
- Alcohol
Result
- Increased ALA synthase activity
Regulation in Bone Marrow
- In erythroid cells, regulation depends mainly on iron availability.
When iron is sufficient
- Heme synthesis increases
- Hemoglobin synthesis proceeds normally
When iron is deficient
- Heme synthesis decreases
Role of Iron in Regulation
- Iron is essential for final step of heme synthesis.
- Without iron, protoporphyrin cannot form heme.
Protoporphyrin IX + Fe2+ → Heme
Role of Vitamin B6
- Vitamin B6 is required for ALA synthase activity.
Deficiency causes
- Reduced heme synthesis
- Sideroblastic anemia
Effect of Lead Poisoning
- Lead inhibits important enzymes:
- ALA dehydratase
- Ferrochelatase
Result
- Heme synthesis decreases
- Anemia develops
Disorder of Heme Biosynthesis
- Disorders of heme biosynthesis occur when one or more enzymes involved in the heme synthesis pathway become deficient or inhibited.
- These disorders lead to accumulation of intermediate compounds of porphyrin metabolism.
- The accumulated substances are toxic and produce clinical symptoms.
- The major disorders are called porphyrias.
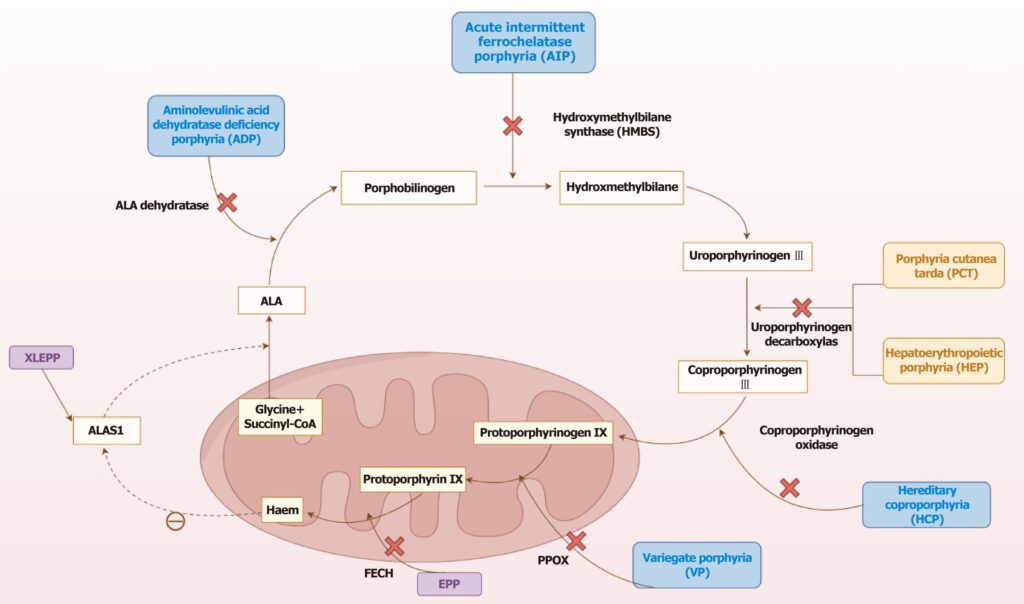
Porphyrias
- Porphyrias are inherited or acquired disorders caused by deficiency of specific enzymes of heme synthesis.
- Because of enzyme deficiency, intermediates before the blocked step accumulate in tissues, blood, urine, or stool.
Common Types of Porphyrias
1. Acute Intermittent Porphyria
- Caused by deficiency of porphobilinogen deaminase
- Accumulation of:
- δ-Aminolevulinic acid (ALA)
- Porphobilinogen
Clinical features
- Severe abdominal pain
- Vomiting
- Constipation
- Neurological symptoms
- Mental disturbances
Important point
- Urine becomes dark on standing
2. Congenital Erythropoietic Porphyria
- Caused by deficiency of uroporphyrinogen III synthase
Clinical features
- Extreme photosensitivity
- Red discoloration of urine
- Skin blistering
- Red coloured teeth
3. Porphyria Cutanea Tarda
- Caused by deficiency of uroporphyrinogen decarboxylase
- Most common porphyria
Clinical features
- Photosensitivity
- Fragile skin
- Blister formation
- Hyperpigmentation
4. Hereditary Coproporphyria
- Caused by deficiency of coproporphyrinogen oxidase
Clinical features
- Abdominal pain
- Neurological symptoms
- Photosensitivity
5. Variegate Porphyria
- Caused by deficiency of protoporphyrinogen oxidase
Clinical features
- Skin lesions
- Neurological attacks
6. Erythropoietic Protoporphyria
- Caused by deficiency of ferrochelatase
Clinical features
- Photosensitivity
- Burning sensation in sunlight
Lead Poisoning and Heme Biosynthesis
Lead inhibits ALA dehydratase and ferrochelatase
- Lead inhibits two important enzymes:
- ALA dehydratase
- Ferrochelatase
Result
- Decreased heme synthesis
- Anaemia develops
Clinical features
- Abdominal pain
- Irritability
- Peripheral neuropathy
- Basophilic stippling of RBCs
Sideroblastic Anaemia
- Caused by defective incorporation of iron into heme
Causes
- Vitamin B6 deficiency
- Defect in ALA synthase
Result
- Iron accumulates in mitochondria
Breakdown of Hemoglobin
- Hemoglobin breakdown occurs when old red blood cells are destroyed after completing their lifespan.
- The average life span of a red blood cell is 120 days.
- Breakdown mainly occurs in the reticuloendothelial system, especially in:
- Spleen
- Liver
- Bone marrow
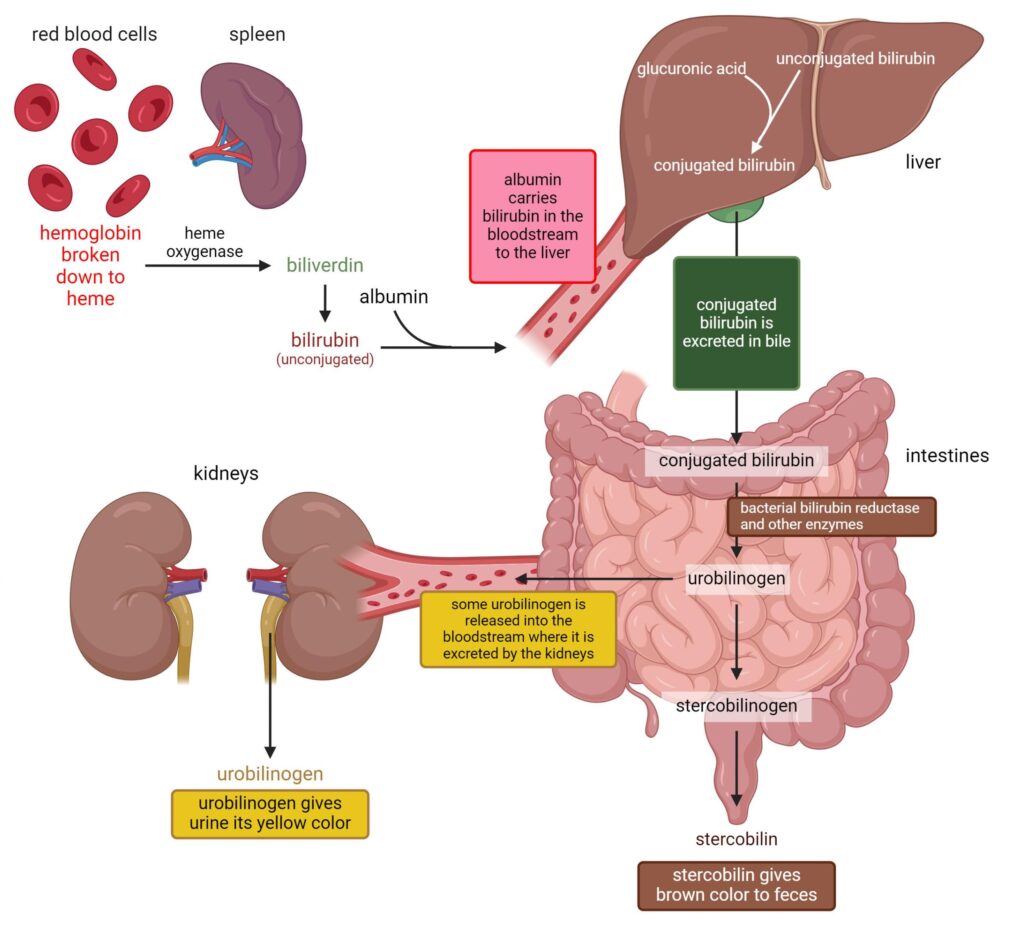
Step 1: Destruction of Old Red Blood Cells
- Old or damaged RBCs are engulfed by macrophages.
- Hemoglobin is released from red blood cells.
Step 2: Separation of Hemoglobin into Globin and Heme
Hemoglobin → Globin + Heme
Fate of Globin
- Globin is the protein part of hemoglobin.
- It is broken down into amino acids.
- Amino acids are reused for synthesis of new proteins.
Fate of Heme
- Heme is the non-protein iron-containing part.
- It undergoes further degradation.
Step 3: Removal of Iron from Heme
- Iron is removed from heme.
- Released iron is transported by transferrin.
- Iron is stored as:
- Ferritin
- Hemosiderin
Importance
- Iron is reused for new hemoglobin synthesis.
Step 4: Formation of Biliverdin
Heme → Biliverdin
- Enzyme: Heme oxygenase
- The porphyrin ring opens.
- Green coloured biliverdin is formed.
- Carbon monoxide (CO) is also released.
Step 5: Formation of Bilirubin
Biliverdin→Bilirubin
- Enzyme: Biliverdin reductase
- Biliverdin is reduced to bilirubin.
- Bilirubin is yellow coloured.
Step 6: Transport of Unconjugated Bilirubin
- Unconjugated bilirubin is insoluble in water.
- It binds with albumin in blood.
- Albumin carries bilirubin to liver.
Step 7: Conjugation in Liver
Bilirubin + Glucuronic acid → Conjugated bilirubin
- Enzyme: UDP-glucuronyl transferase
- Bilirubin combines with glucuronic acid.
- Conjugated bilirubin becomes water soluble.
Step 8: Excretion in Bile
- Conjugated bilirubin is secreted into bile.
- It reaches intestine through bile duct.
Step 9: Formation of Urobilinogen in Intestine
- Intestinal bacteria convert bilirubin into urobilinogen.
Fate of Urobilinogen
- Part is reabsorbed into blood
- Part goes to kidney and forms urobilin
- Most is converted into stercobilin
Final Pigments
- Urobilin gives yellow colour to urine
- Stercobilin gives brown colour to feces
Hyperbilirubinemias
- Hyperbilirubinemia means abnormally increased bilirubin concentration in blood.
- Bilirubin is a yellow pigment formed during the breakdown of heme from hemoglobin.
- Normally bilirubin produced from old red blood cells is transported to liver, conjugated, and excreted through bile.
- When this process is disturbed at any stage, bilirubin accumulates in blood and tissues.
- Increased bilirubin causes jaundice, characterized by yellow discoloration of:
- Skin
- Sclera
- Mucous membranes
Normal Serum Bilirubin Level
- Normal total bilirubin: 0.2–1.2 mg/dL
- Direct (conjugated) bilirubin: 0–0.3 mg/dL
- Indirect (unconjugated) bilirubin: 0.2–0.8 mg/dL
Types of Hyperbilirubinemias
Hyperbilirubinemia is divided into:
- Unconjugated (Indirect) Hyperbilirubinemia
- Conjugated (Direct) Hyperbilirubinemia
1. Unconjugated Hyperbilirubinemia
- In this condition, unconjugated bilirubin increases in blood before liver conjugation.
- Unconjugated bilirubin is:
- Water insoluble
- Bound to albumin
- Cannot pass into urine
Causes of Unconjugated Hyperbilirubinemia
A. Increased Bilirubin Production
Occurs due to excessive hemolysis.
Causes
- Hemolytic anemia
- Malaria
- Mismatched blood transfusion
- Sickle cell disease
- Thalassemia
Mechanism
- Large RBC destruction increases bilirubin production beyond liver capacity.
B. Defective Hepatic Uptake
Liver fails to take bilirubin efficiently.
Causes
- Certain drugs
- Liver dysfunction
C. Defective Conjugation in Liver
Occurs when liver enzyme is deficient.
Important enzyme
UDP-glucuronyl transferase
Disorders due to conjugation defect
Gilbert Syndrome
- Mild inherited reduction in UDP-glucuronyl transferase activity
- Common and usually harmless
Features
- Mild jaundice during stress or fasting
Crigler–Najjar Syndrome Type I
- Complete absence of UDP-glucuronyl transferase
Features
- Severe neonatal jaundice
- Very high unconjugated bilirubin
- Kernicterus may occur
Crigler–Najjar Syndrome Type II
- Partial deficiency of enzyme
Features
- Less severe than Type I
Neonatal Physiological Jaundice

- Newborn liver enzyme system is immature.
- Conjugation capacity is low.
Result
- Unconjugated bilirubin rises after birth
Danger
- Severe elevation may cause kernicterus (bilirubin deposition in brain)
Features of Unconjugated Hyperbilirubinemia
- Bilirubin absent in urine
- Stool colour normal
- No dark urine
2. Conjugated Hyperbilirubinemia
- In this type, bilirubin is conjugated normally but cannot be excreted properly.
- Conjugated bilirubin is:
- Water soluble
- Appears in urine
Causes of Conjugated Hyperbilirubinemia
A. Hepatocellular Disease
Liver cells are damaged.
Causes
- Viral hepatitis
- Alcoholic liver disease
- Cirrhosis
- Drug-induced liver injury
- Liver metastasis
Mechanism
- Liver conjugates bilirubin but secretion into bile decreases.
B. Obstruction of Bile Flow (Obstructive Jaundice)
- Gallstones
- Carcinoma head of pancreas
- Biliary stricture
- Primary sclerosing cholangitis
Mechanism
- Conjugated bilirubin refluxes back into blood.
Features of Conjugated Hyperbilirubinemia
- Bilirubin present in urine
- Dark urine
- Clay coloured stool
- Itching may occur
Classification According to Site of Defect
Pre-hepatic Hyperbilirubinemia
- Excess RBC destruction
Bilirubin type
- Unconjugated bilirubin increased
Hepatic Hyperbilirubinemia
- Liver cell dysfunction
Bilirubin type
- Mixed rise of conjugated and unconjugated bilirubin
Post-hepatic Hyperbilirubinemia
- Bile duct obstruction
Bilirubin type
- Conjugated bilirubin increased
Inherited Conjugated Hyperbilirubinemias
Dubin–Johnson Syndrome
- Defect in transport of conjugated bilirubin into bile
Features
- Conjugated bilirubin rises
- Liver appears black
Rotor Syndrome
- Similar to Dubin–Johnson syndrome
- No liver pigmentation
Clinical Importance
- Hyperbilirubinemia helps diagnose:
- Hemolysis
- Liver disease
- Biliary obstruction
- Serum bilirubin estimation is an important liver function test.
Severe Complication
Kernicterus
- Unconjugated bilirubin crosses blood-brain barrier in newborns
- Causes brain damage
Transport of Oxygen by Hemoglobin
- Hemoglobin is the principal oxygen-carrying protein of blood.
- It transports oxygen from the lungs to peripheral tissues where oxygen is required for cellular metabolism.
- Oxygen transport is essential because every cell needs oxygen for aerobic respiration and ATP production.
- About 97% of oxygen in blood is transported by hemoglobin, while only about 3% remains dissolved in plasma.
Oxygen Loading in Lungs
- In the lungs, oxygen partial pressure (pO₂) is high.
- Oxygen diffuses from alveoli into blood.
- Hemoglobin binds oxygen and forms oxyhemoglobin.
Hb+O2⇌HbO2
- At pulmonary capillaries, hemoglobin becomes almost fully saturated.
Oxygen Unloading in Tissues
- In tissues, oxygen partial pressure is low because oxygen is continuously used by cells.
- Oxyhemoglobin releases oxygen, which enters tissues for metabolism.
Oxygen Dissociation Curve
- Oxygen dissociation curve is a graphical representation showing the relationship between:
- Partial pressure of oxygen (pO₂)
- Percentage saturation of hemoglobin
- The curve is characteristically sigmoid (S-shaped).
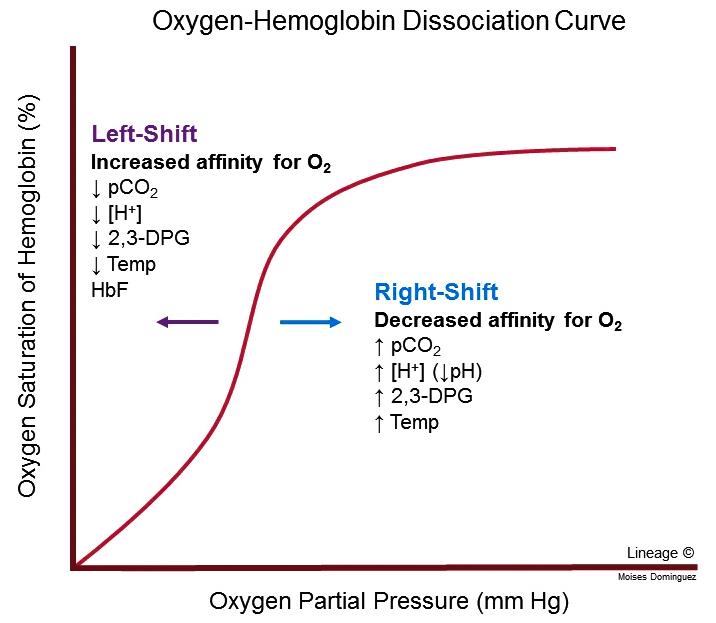
Curve is Sigmoid
- The sigmoid shape is due to cooperative binding of oxygen.
- Binding of one oxygen molecule increases affinity for subsequent oxygen molecules.
Functional Importance of Different Parts of ODC
Upper flat portion
- Represents oxygen loading in lungs
- Even moderate fall in alveolar pO₂ does not greatly reduce saturation
Steep portion
- Represents oxygen unloading in tissues
- Small fall in pO₂ causes large oxygen release
P50 Value
- P50 is the pO₂ at which hemoglobin is 50% saturated.
- Normal P50 = approximately 26 mmHg
Importance
- Indicates oxygen affinity of hemoglobin
Heme-Heme Interaction and Cooperativity
- Hemoglobin exhibits positive cooperativity.
- The first oxygen binds with some difficulty because deoxyhemoglobin is in T form.
- Once first oxygen binds, structural changes occur that make second oxygen binding easier.
Sequential Binding
- First oxygen binds slowly
- Second oxygen binds more rapidly
- Third oxygen binds even faster
- Fourth oxygen binds with highest affinity
Structural Basis
- Binding of oxygen breaks some ionic and hydrogen bonds between globin chains.
- Hemoglobin shifts from:
T form (tense form)
- Low oxygen affinity
R form (relaxed form)
- High oxygen affinity
Importance
- Ensures rapid oxygen loading in lungs
- Ensures efficient unloading in tissues
Effect of pH and pCO₂
- Tissue metabolism produces carbon dioxide and hydrogen ions.
- Both lower oxygen affinity of hemoglobin.
Effect of Increased H⁺
- Hydrogen ions bind globin chains.
- Stabilize T form of hemoglobin.
Result
- Oxygen release increases
Effect of Increased pCO₂
- CO₂ binds terminal amino groups of globin.
Forms carbaminohemoglobin
- Carbaminohemoglobin decreases oxygen affinity.
Overall Effect
- Active tissues with high CO₂ and acidity receive more oxygen.
The Bohr Effect
- The Bohr effect describes the influence of hydrogen ion concentration (H⁺) and carbon dioxide (CO₂) on the oxygen-binding capacity of hemoglobin.
- When tissues become metabolically active, they produce more carbon dioxide and acids. This lowers the pH of blood in tissues.
- Under these conditions, hemoglobin loses its affinity for oxygen and releases more oxygen to tissues.
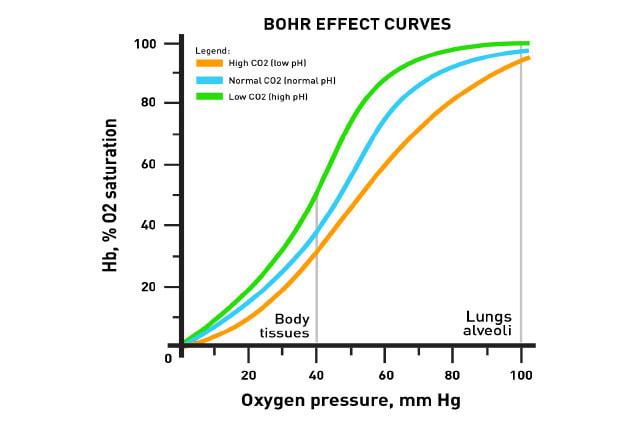
Mechanism of the Bohr Effect
- Carbon dioxide produced in tissues enters red blood cells.
- Inside RBCs, carbon dioxide combines with water.
CO2+H2O→H2CO3→H++HCO3−
- Carbonic acid formed dissociates into:
- Hydrogen ions (H⁺)
- Bicarbonate ions (HCO₃⁻)
- Hydrogen ions bind to globin chains of hemoglobin.
- This stabilizes the T form (tense form) of hemoglobin, which has lower oxygen affinity.
Effect on Oxygen Dissociation Curve
- The oxygen dissociation curve shifts to the right.
- A right shift means hemoglobin releases oxygen more easily.
Physiological Importance
- Active tissues receive more oxygen.
- This is especially important during:
- Exercise
- Fever
- Increased muscular activity
The Chloride Shift
- The chloride shift is the exchange of bicarbonate and chloride ions across the red blood cell membrane.
- It occurs during transport of carbon dioxide from tissues to lungs.
- It is also called the Hamburger phenomenon.
Mechanism in Tissues
- Carbon dioxide enters red blood cells.
- Carbonic anhydrase converts carbon dioxide into carbonic acid.
- Carbonic acid dissociates into bicarbonate and hydrogen ions.
CO2 + H2O → H2CO3 → H+ + HCO3−
- Bicarbonate ions leave RBC and enter plasma.
- To maintain electrical neutrality, chloride ions enter RBC.
Mechanism in Lungs
- In lungs, bicarbonate re-enters RBC.
- Chloride leaves RBC.
- Carbonic acid reforms and decomposes into CO₂ and water.
- CO₂ is exhaled.
Importance of Chloride Shift
- Maintains ionic balance in red blood cells
- Helps efficient transport of CO₂ in blood
- Supports acid-base balance
Effect of Temperature
- Temperature significantly affects oxygen binding by hemoglobin.
- Increased temperature reduces oxygen affinity of hemoglobin.
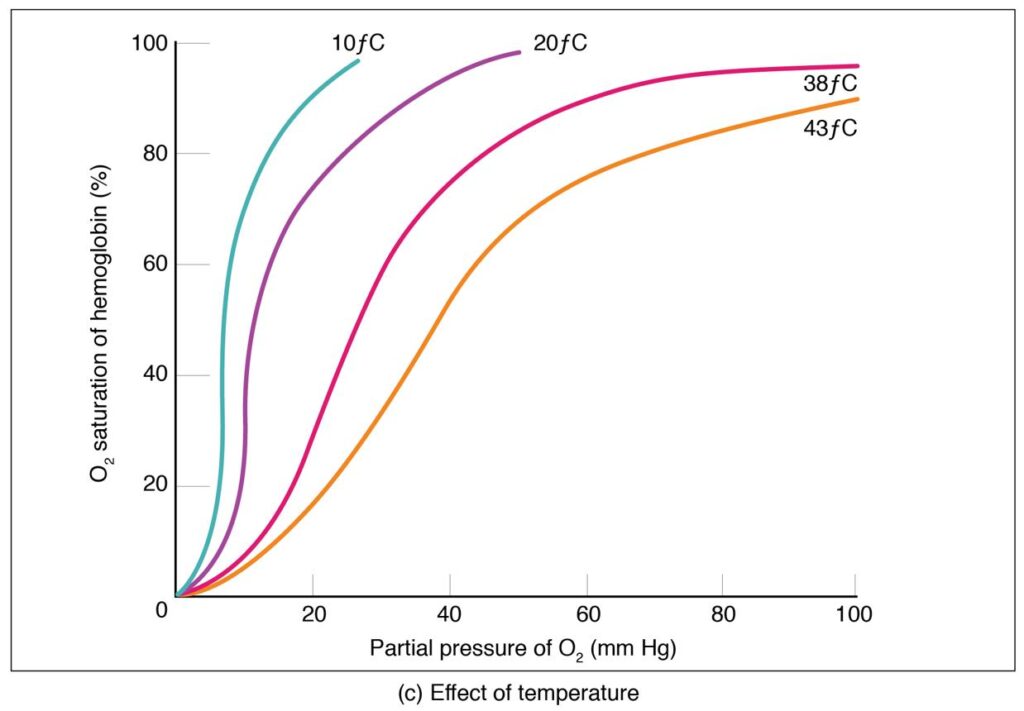
Mechanism
- Higher temperature weakens the bond between oxygen and hemoglobin.
- Oxygen is released more easily to tissues.
- Oxygen dissociation curve shifts to the right.
Situations Where Temperature Increases
- Exercise
- Fever
- Active muscle metabolism
Physiological Importance
- Working muscles produce heat and require more oxygen.
- Increased temperature helps oxygen delivery exactly where needed.
Effect of 2,3-BPG
- 2,3-BPG (2,3-bisphosphoglycerate) is an important regulator of oxygen transport by hemoglobin.
- It is present inside red blood cells and is formed during glycolysis.
- 2,3-BPG controls how easily hemoglobin releases oxygen to tissues.
- Its main role is to reduce the affinity of hemoglobin for oxygen, thereby promoting oxygen unloading in peripheral tissues.
Formation of 2,3-BPG
- 2,3-BPG is produced in erythrocytes from glycolytic intermediates through the Rapoport–Luebering shunt.
- It is formed from 1,3-bisphosphoglycerate during glycolysis.
- Because mature red blood cells do not have mitochondria, glycolysis is their main source of energy, and 2,3-BPG is produced as part of this pathway.
Site of Action
- 2,3-BPG acts directly on deoxyhemoglobin.
- It binds in the central cavity between the two beta chains of hemoglobin.
Mechanism of Action
- 2,3-BPG binds strongly to deoxygenated hemoglobin (T form).
- It stabilizes the T form (tense form) of hemoglobin.
- T form has lower affinity for oxygen.
Result
- Hemoglobin does not hold oxygen tightly
- Oxygen is released more easily into tissues
Conditions that Increase 2,3-BPG
- High altitude
- Chronic hypoxia
- Severe anemia
- Chronic lung disease
- Congestive heart failure
Transport of Carbon Dioxide
- Carbon dioxide (CO₂) is continuously produced in body tissues as the final product of aerobic metabolism.
- Every actively metabolizing cell generates carbon dioxide during oxidation of carbohydrates, fats, and proteins.
- Since accumulation of CO₂ increases acidity, it must be rapidly transported to the lungs and excreted.
- Blood performs this function efficiently by carrying carbon dioxide from tissues to lungs in different chemical forms.
Major Forms of Carbon Dioxide Transport
Carbon dioxide is transported in blood in three main forms:
- Dissolved form
- Bicarbonate form (Isohydric transport)
- Carbamino compounds
Relative Distribution
- About 5–10% as dissolved CO₂
- About 60–70% as bicarbonate
- About 20–25% as carbamino compounds
Among these, bicarbonate transport is the major mechanism.
Dissolved Form
- A small portion of carbon dioxide is transported directly dissolved in plasma.
- Carbon dioxide is about 20 times more soluble in plasma than oxygen.
- Because of this greater solubility, some carbon dioxide remains physically dissolved without chemical change.
Amount Transported
- Approximately 5–10% of total carbon dioxide is carried in dissolved form.
Physiological Importance
- Dissolved carbon dioxide contributes directly to blood pCO₂ (partial pressure of carbon dioxide).
- Respiratory centers in brain respond mainly to pCO₂ levels.
- Thus dissolved CO₂ plays an important role in respiratory regulation.
In Tissues
- Carbon dioxide concentration is high in tissues due to metabolism.
- It diffuses into capillary blood because tissue pCO₂ is greater than blood pCO₂.
In Lungs
- Blood reaching lungs has higher pCO₂ than alveolar air.
- Dissolved carbon dioxide diffuses into alveoli and is expired.
Isohydric Transport of Carbon Dioxide
- Isohydric transport means carbon dioxide is transported mainly as bicarbonate while blood pH remains nearly constant.
- This is the most important mechanism of carbon dioxide transport.
Entry of Carbon Dioxide into RBC
- Carbon dioxide diffuses from tissues into red blood cells.
- Inside RBCs, carbon dioxide rapidly combines with water.
CO2 + H2O → H2CO3 → H+ + HCO3
Role of Carbonic Anhydrase
- The enzyme carbonic anhydrase present in RBCs catalyzes this reaction very rapidly.
- Without this enzyme, reaction would be slow.
Formation of Carbonic Acid
- Carbon dioxide combines with water to form carbonic acid (H₂CO₃).
Dissociation of Carbonic Acid
- Carbonic acid dissociates into:
- Hydrogen ion (H⁺)
- Bicarbonate ion (HCO₃⁻)
Role of Hemoglobin in Buffering
- Hemoglobin immediately binds hydrogen ions.
Importance
- Prevents fall in blood pH
- Maintains acid-base balance
Why Called Isohydric Transport?
- Because carbon dioxide is transported without major change in blood pH.
Chloride Shift During Isohydric Transport
- Bicarbonate formed inside RBC diffuses into plasma.
- To maintain electrical neutrality, chloride ions enter RBC.
This is called chloride shift.
Importance
- About 60–70% of total carbon dioxide is transported by this mechanism.
Carriage as Carbamino-Hemoglobin
- Carbon dioxide also binds directly with proteins in blood.
- Hemoglobin is the major protein involved.
Fetal Hemoglobin
- Fetal hemoglobin (HbF) is the major hemoglobin present during fetal life.
- It is specially designed to supply oxygen efficiently from maternal blood to the fetus through the placenta.
- During intrauterine life, fetal tissues depend completely on maternal oxygen supply, therefore fetal hemoglobin has a higher affinity for oxygen than adult hemoglobin.
Structure of Fetal Hemoglobin
- Fetal hemoglobin contains four globin chains.
Composition
HbF = α2γ2
- It contains:
- 2 alpha (α) chains
- 2 gamma (γ) chains
Difference from Adult Hemoglobin
Adult hemoglobin (HbA) contains:
HbA=α2β2
- In fetal hemoglobin, beta chains are replaced by gamma chains.
Period of Presence
- HbF is the major hemoglobin from fetal life until birth.
- It constitutes about 70–90% of total hemoglobin in fetus.
- After birth, HbF gradually decreases.
Replacement After Birth
- Gamma chain synthesis decreases after birth.
- Beta chain synthesis increases.
- Adult hemoglobin gradually replaces HbF.
Time of Replacement
- By about 6 months of age, most HbF is replaced by HbA.
Measurement of HbF
- HbF estimation is useful in:
- Thalassemia
- Sickle cell disease
- Hemolytic disorders
Hemoglobin A2
- Hemoglobin A₂ (HbA₂) is a minor normal hemoglobin present in adult blood.
- Although it is present in small quantity, it is clinically important because its level changes in some blood disorders, especially thalassemia.
Structure of Hemoglobin A₂
- Hemoglobin A₂ contains four globin chains.
Composition
HbA2=α2δ2
- It contains:
- 2 alpha (α) chains
- 2 delta (δ) chains
Comparison with Adult Hemoglobin
Adult major hemoglobin (HbA) contains:
HbA=α2β2
- In HbA₂, beta chains are replaced by delta chains.
Percentage in Normal Adults
- HbA₂ normally forms about 2–3% of total adult hemoglobin.
- Usually its concentration remains less than 3.5%.
Time of Appearance
- HbA₂ appears after birth as fetal hemoglobin decreases.
- Its level gradually reaches adult concentration during infancy.
Site of Synthesis
- HbA₂ is synthesized in bone marrow during erythropoiesis.
Functional Properties
- HbA₂ performs oxygen transport similar to HbA.
- Its oxygen affinity is slightly different but functionally very similar to adult hemoglobin.
Clinical Importance of HbA₂
- HbA₂ estimation is important in diagnosis of β-thalassemia trait.
In β-Thalassemia Trait
- Beta chain synthesis decreases.
- Delta chain production relatively increases.
Result
- HbA₂ level becomes elevated.
Diagnostic Value
- HbA₂ more than 3.5% strongly suggests β-thalassemia trait.
In Iron Deficiency Anemia
- HbA₂ may remain normal or slightly reduced.
This helps differentiate iron deficiency anemia from thalassemia trait.
Laboratory Detection
- HbA₂ is measured by:
- Hemoglobin electrophoresis
- High-performance liquid chromatography (HPLC)
Hemoglobin Derivatives
- Hemoglobin derivatives are various forms of hemoglobin produced when hemoglobin combines with gases, undergoes oxidation, or reacts with different chemical substances.
- These derivatives differ in structure, colour, oxygen affinity, and physiological significance.
- Some derivatives are formed normally during respiration, while others are abnormal and may impair oxygen delivery to tissues.
- Understanding hemoglobin derivatives is important because they help explain normal gas transport as well as many clinical disorders such as carbon monoxide poisoning, methemoglobinemia, and diabetes mellitus.
1. Oxyhemoglobin

- Oxyhemoglobin is the normal physiological derivative formed when oxygen binds reversibly with hemoglobin in the lungs.
- Oxygen binds specifically to ferrous iron (Fe²⁺) present in each heme group.
- Because one hemoglobin molecule contains four heme groups, one hemoglobin molecule can bind four oxygen molecules.
Hb+O2⇌HbO2
Structural Characteristics
- Iron remains in ferrous state (Fe²⁺) during oxygenation.
- Oxygen binding is reversible.
- No oxidation of iron occurs during normal oxygen transport.
Colour
- Oxyhemoglobin gives arterial blood its bright red colour.
Formation Site
- Formed mainly in pulmonary capillaries where oxygen partial pressure is high.
Physiological Importance
- Responsible for oxygen transport from lungs to tissues.
- Maintains oxygen supply for aerobic metabolism.
Dissociation in Tissues
- In tissues where oxygen tension is low, oxyhemoglobin dissociates and releases oxygen.
2. Reduced Hemoglobin (Deoxyhemoglobin)
- Reduced hemoglobin is hemoglobin after oxygen has been released to tissues.
- It represents deoxygenated hemoglobin circulating in venous blood.
Structural Characteristics
- Iron remains in ferrous state (Fe²⁺).
- No oxygen molecule is attached.
Colour
- Gives venous blood a dark red colour.
Physiological Role
- Facilitates carbon dioxide transport because deoxyhemoglobin binds CO₂ and H⁺ more readily than oxyhemoglobin.
Clinical Importance
- Increased reduced hemoglobin causes cyanosis when its concentration exceeds about 5 g/dL.
3. Carbaminohemoglobin
- Carbaminohemoglobin is formed when carbon dioxide combines with terminal amino groups of globin chains.
- Carbon dioxide does not bind to heme iron.
Hb − NH2 + CO2 → Hb − NHCOO− + H+
Formation Site
- Mainly formed in peripheral tissues where carbon dioxide concentration is high.
Amount Transported
- About 20–25% of total carbon dioxide is carried as carbaminohemoglobin.
Why Deoxyhemoglobin Binds More CO₂
- Deoxygenated hemoglobin has greater affinity for carbon dioxide.
Importance
- Major secondary mechanism of carbon dioxide transport.
In Lungs
- Oxygenation of hemoglobin releases carbon dioxide.
4. Carboxyhemoglobin
- Carboxyhemoglobin forms when carbon monoxide binds with hemoglobin.
- Carbon monoxide occupies the same binding site used by oxygen.
Hb+CO→HbCOHb + CO \rightarrow HbCO
Affinity for Carbon Monoxide
- Hemoglobin affinity for carbon monoxide is 200–250 times greater than oxygen.
Consequences
- Oxygen cannot bind effectively.
- Tissue hypoxia develops even when blood oxygen content appears normal.
Colour
- Blood becomes cherry red.
Clinical Features
- Headache
- Dizziness
- Confusion
- Unconsciousness
- Respiratory failure
Common Sources
- Vehicle exhaust
- Fire smoke
- Incomplete combustion
Treatment
- 100% oxygen administration
- Hyperbaric oxygen in severe poisoning
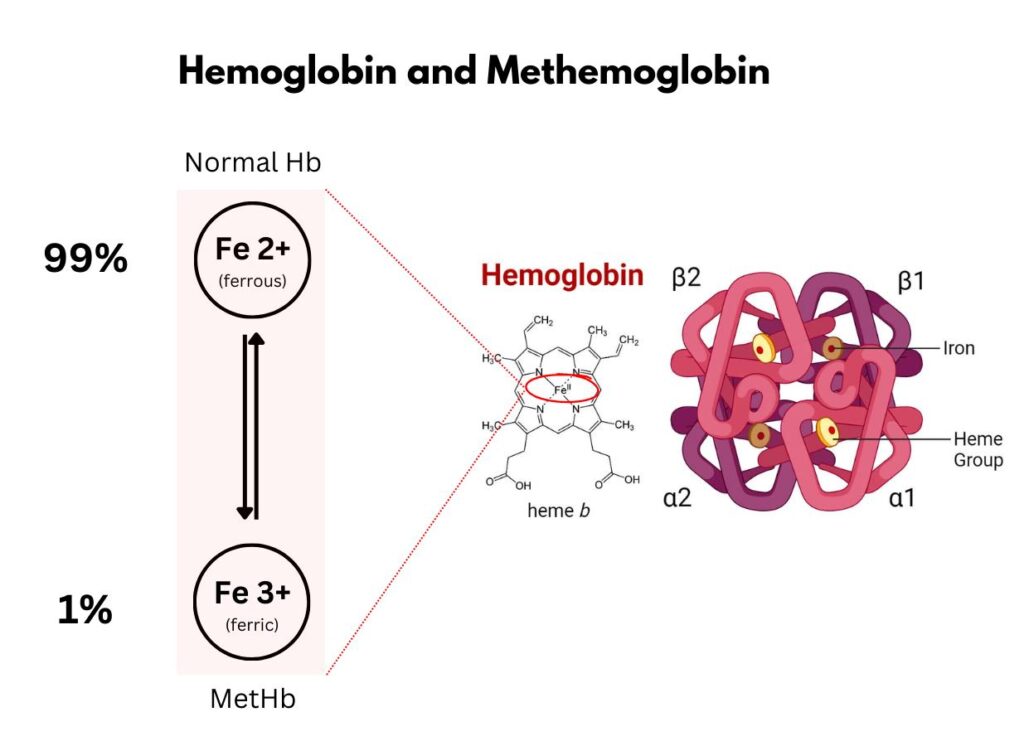
5. Methemoglobin
- Methemoglobin is formed when iron in hemoglobin is oxidized from Fe²⁺ to Fe³⁺.
Fe2+ → Fe3+
Structural Change
- Ferric iron (Fe³⁺) cannot bind oxygen.
Effect
- Oxygen transport capacity decreases significantly.
Causes
- Nitrite poisoning
- Sulfonamides
- Local anesthetics
- Congenital methemoglobin reductase deficiency
Colour
- Blood appears chocolate brown.
Symptoms
- Cyanosis
- Breathlessness
- Fatigue
- Severe hypoxia in high levels
Protective Mechanism
- RBC enzyme methemoglobin reductase converts Fe³⁺ back to Fe²⁺.
Treatment
- Methylene blue in severe cases
6. Sulfhemoglobin
- Sulfhemoglobin forms when sulfur irreversibly binds to hemoglobin.
Nature
- Irreversible derivative
- Cannot carry oxygen efficiently
Causes
- Sulfur-containing drugs
- Hydrogen sulfide exposure
Clinical Features
- Persistent cyanosis
- Blood may appear greenish-dark
Important Difference
- Unlike methemoglobin, sulfhemoglobin cannot be reduced back.
7. Glycosylated Hemoglobin (HbA1c)
- Glycosylated hemoglobin forms by non-enzymatic attachment of glucose to hemoglobin.
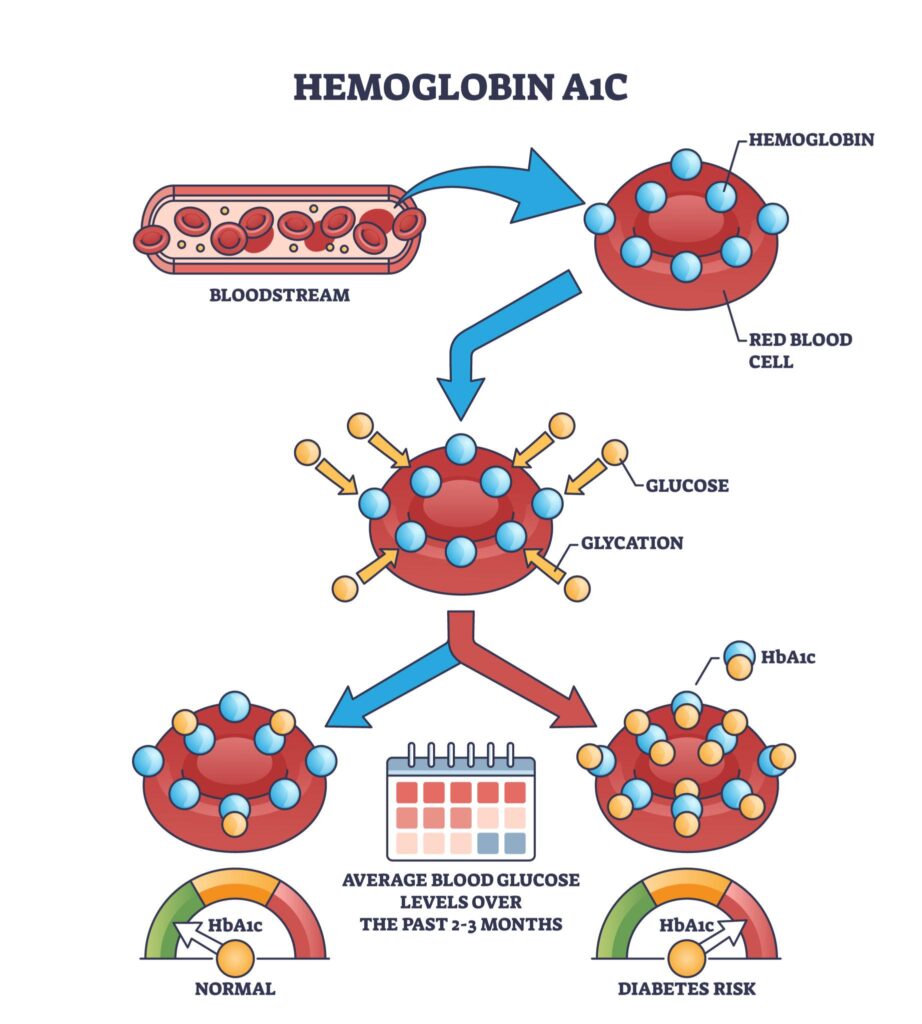
Site of Binding
- Glucose binds mainly to N-terminal valine of beta chain.
Formation
- Slow irreversible reaction during RBC lifespan.
Clinical Importance
- Reflects average blood glucose over previous 8–12 weeks.
Uses
- Diagnosis of diabetes mellitus
- Monitoring long-term glycemic control
Increased In
- Diabetes mellitus
- Poor glycemic control
Hemoglobin (Globin Chain) Variants
- Hemoglobin variants are structurally abnormal hemoglobins produced due to inherited alterations in the amino acid sequence of globin chains.
- These abnormalities arise because of mutations in genes that encode alpha, beta, gamma, or delta globin chains.
- In most clinically important cases, mutation affects the beta globin chain, because beta chain synthesis becomes dominant after birth.
- A single amino acid substitution may significantly alter the physical and functional properties of hemoglobin, including:
- Solubility
- Stability
- Oxygen affinity
- Molecular charge
- Polymerization tendency
- Such structural abnormalities are called hemoglobinopathies.
Molecular Basis of Hemoglobin Variants
- Most hemoglobin variants occur due to point mutation in globin gene DNA.
- A point mutation changes one nucleotide in DNA sequence.
- This leads to change in codon during translation.
- As a result, one amino acid in globin chain is replaced by another amino acid.
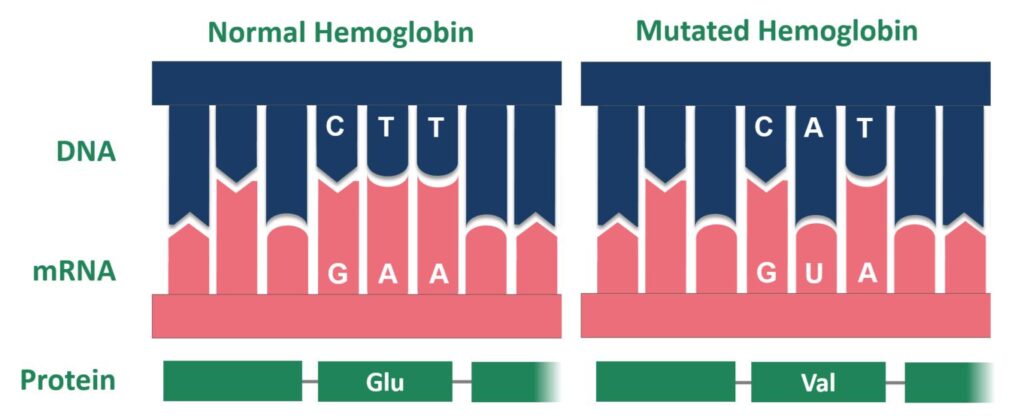
Consequences of Amino Acid Substitution
The substituted amino acid may alter:
- Charge of hemoglobin molecule
- Surface hydrophobicity
- Solubility in deoxygenated state
- Molecular folding
- Interaction between hemoglobin molecules
Result
- Abnormal RBC shape
- Reduced RBC lifespan
- Hemolysis
- Tissue hypoxia
Classification of Hemoglobin Variants
Hemoglobin variants are broadly classified into:
- Variants due to beta chain mutation
- Variants due to alpha chain mutation
- Variants with altered oxygen affinity
- Variants producing unstable hemoglobin
1. Hemoglobin S (HbS)
- Hemoglobin S is the most clinically important hemoglobin variant.
- It is responsible for sickle cell disease.
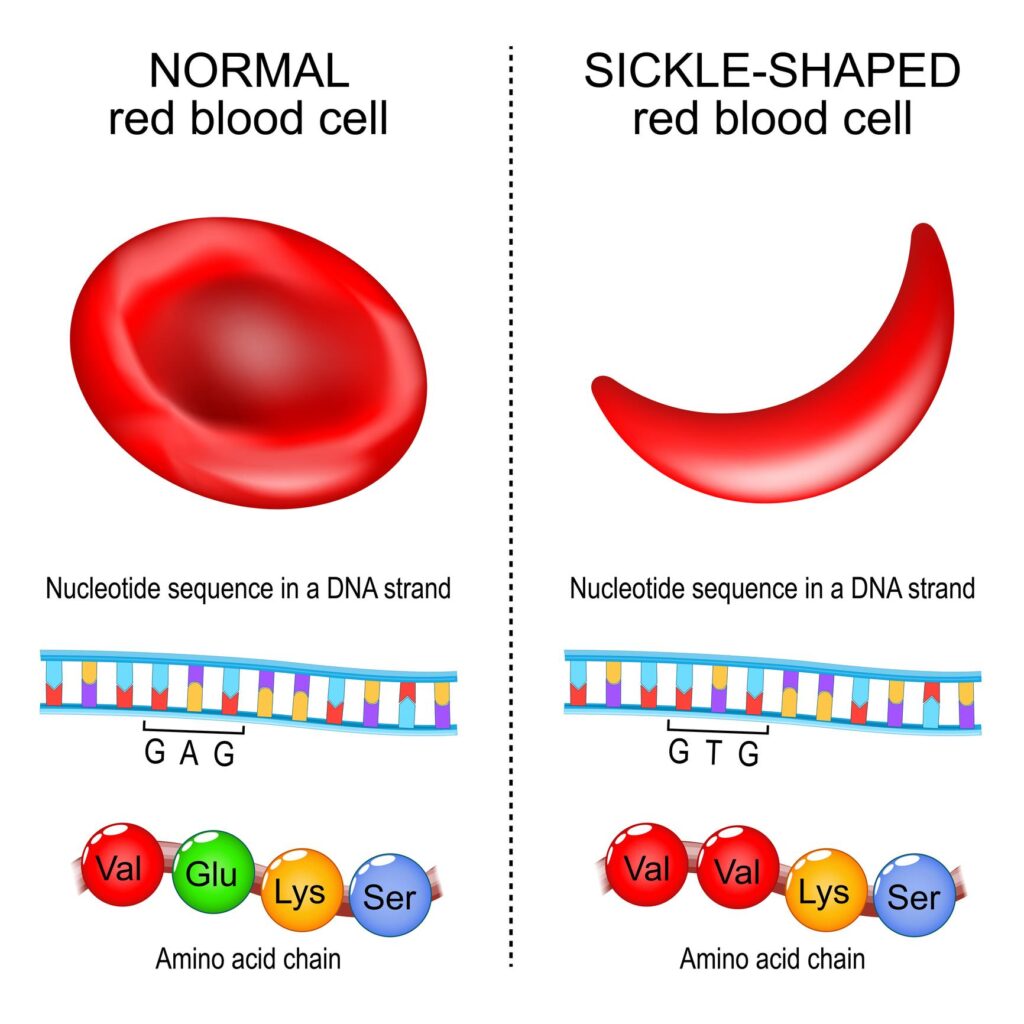
Molecular Defect
β6 Glu→Val
- At position 6 of beta globin chain:
- Glutamic acid is replaced by valine.
Why This Mutation is Important
- Glutamic acid is hydrophilic.
- Valine is hydrophobic.
Result
- Deoxygenated HbS molecules stick together.
Polymerization
- In low oxygen tension, HbS polymerizes into long fibers.
Effect on Red Blood Cells
- RBC becomes elongated and sickle shaped
- Membrane rigidity increases
- Cells become fragile
Consequences
- Hemolysis
- Microvascular obstruction
- Tissue ischemia
Clinical Features
- Chronic hemolytic anemia
- Painful vaso-occlusive crises
- Bone pain
- Splenomegaly
- Jaundice
- Leg ulcers
- Stroke in severe cases
Sickling Triggered By
- Hypoxia
- Acidosis
- Dehydration
- Infection
Genetic Forms
Heterozygous (Sickle cell trait)
- HbAS
- Usually asymptomatic
Homozygous (Sickle cell disease)
- HbSS
- Severe disease
2. Hemoglobin C (HbC)

- HbC is another beta chain structural variant.
Molecular Defect
β6 Glu→Lys
- Glutamic acid replaced by lysine at beta chain position 6.
Effect of Mutation
- Lysine changes molecular charge
- Hemoglobin becomes less soluble
Cellular Effect
- Intracellular crystals may form
- RBC becomes rigid
Clinical Features
- Mild chronic hemolytic anemia
- Splenomegaly
- Target cells in peripheral smear
Severity
- Usually milder than sickle cell disease
3. Hemoglobin D (HbD)
- HbD is another beta chain variant usually clinically mild.
Molecular Defect
β121 Glu→Gln
- Glutamic acid replaced by glutamine at position 121.
Clinical Features
- Usually asymptomatic
- Mild hemolysis may occur
Importance
- Significant if inherited with HbS.
4. Hemoglobin E (HbE)
- HbE is very common in Southeast Asia.
Molecular Defect
β26 Glu→Lys
- Glutamic acid replaced by lysine at beta chain position 26.
Effect
- Structural abnormality plus reduced beta chain synthesis
Clinical Features
- Mild anemia
- Microcytosis
Importance
- Severe disease when combined with beta-thalassemia
5. Hemoglobin M (HbM)
- HbM is a variant causing persistent methemoglobinemia.
Defect
- Amino acid substitution stabilizes ferric iron (Fe³⁺).
Result
- Iron remains oxidized
- Oxygen cannot bind properly
Clinical Feature
- Cyanosis present from birth
Alpha Chain Variants
- Alpha chain variants are less common than beta chain variants.
Examples
- Hb G
- Hb Q
Clinical Significance
- Usually mild
- Sometimes discovered incidentally
Variants with Altered Oxygen Affinity
Some variants alter oxygen binding rather than RBC shape.
High oxygen affinity variants
- Oxygen released poorly to tissues
- Secondary polycythemia develops
Low oxygen affinity variants
- Oxygen released easily
- Usually mild cyanosis
Laboratory Detection of Hemoglobin Variants
- Hemoglobin electrophoresis
- High-performance liquid chromatography (HPLC)
- DNA analysis
- Molecular genetic testing
Clinical Importance
- Early diagnosis of hemoglobinopathies
- Carrier detection
- Prenatal diagnosis
- Genetic counseling
Thalassemias
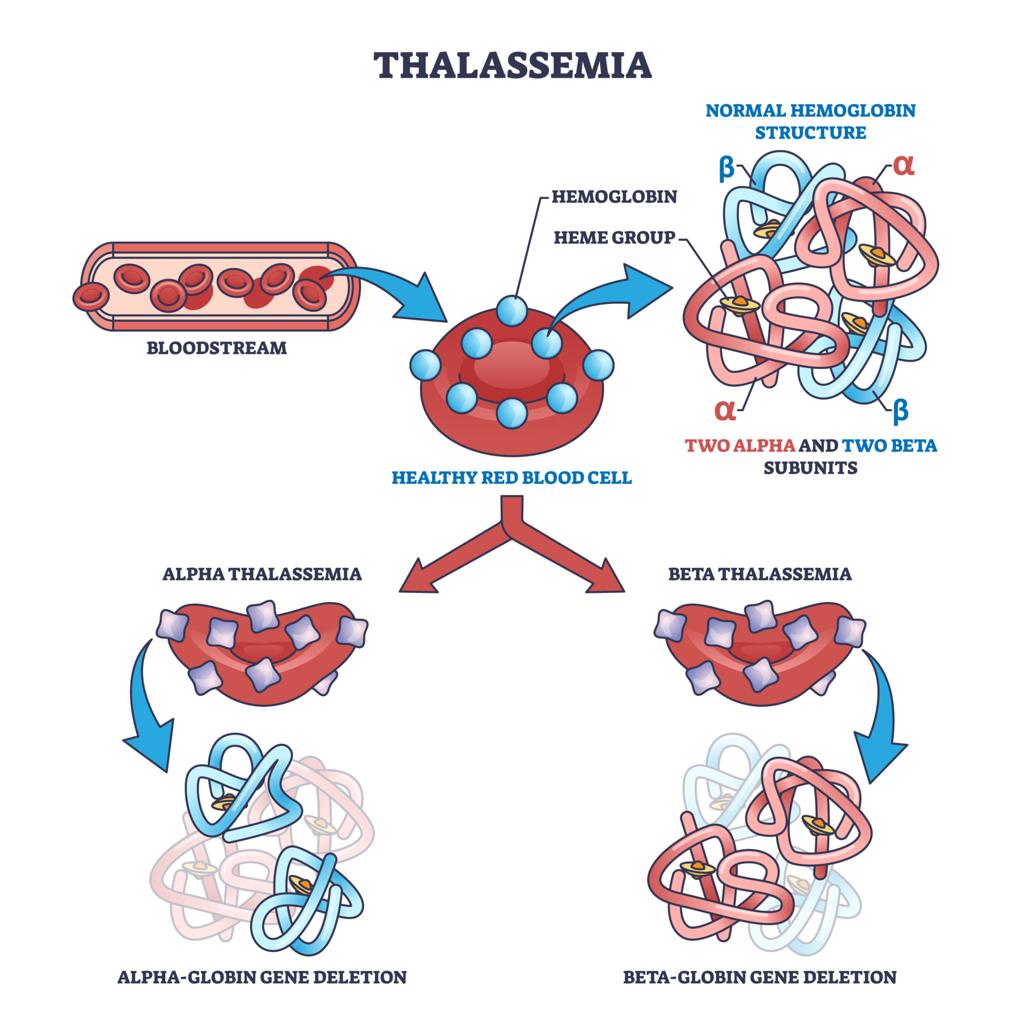
- Thalassemias are inherited disorders characterized by decreased or absent synthesis of one or more globin chains of hemoglobin.
- Unlike hemoglobin variants, where globin structure is abnormal, in thalassemia the globin chain structure is normal but the amount produced is reduced.
- Because globin chain production becomes imbalanced, normal hemoglobin synthesis is disturbed and red blood cells become defective.
- Thalassemias produce chronic hemolytic anemia of varying severity.
- Hemoglobin requires balanced production of alpha and beta globin chains.
- If one chain is reduced, excess unmatched chains accumulate.
Result of Chain Imbalance
- Unpaired globin chains precipitate inside RBC precursors.
- Red cell membrane becomes damaged.
- Ineffective erythropoiesis develops.
- Premature destruction of RBC occurs.
Consequences
- Microcytic hypochromic anemia
- Hemolysis
- Bone marrow expansion
- Increased iron absorption
Classification of Thalassemias
Thalassemias are classified according to affected globin chain:
- Alpha thalassemia
- Beta thalassemia
Alpha Thalassemia
- Alpha thalassemia occurs due to reduced or absent synthesis of alpha globin chains.
- Alpha globin genes are located on chromosome 16.
- Each individual normally has four alpha globin genes (two on each chromosome).
Cause
- Usually caused by gene deletion.
Severity Depends on Number of Deleted Genes
1 Gene Deletion
- Silent carrier state
Features
- Usually asymptomatic
- Normal hemoglobin level
2 Gene Deletion
- Alpha thalassemia trait
Features
- Mild microcytic anemia
3 Gene Deletion
- Hemoglobin H disease
β4=HbH
Features
- Excess beta chains form tetramers
- Moderate to severe hemolytic anemia
- Splenomegaly
4 Gene Deletion
- Hydrops fetalis
γ4=Hb Bart′s
Features
- No alpha chain synthesis
- Severe fetal hypoxia
- Usually fatal before or shortly after birth
Beta Thalassemia
- Beta thalassemia occurs due to reduced or absent beta globin synthesis.
- Beta globin gene is located on chromosome 11.
Cause
- Usually point mutation affecting transcription or translation.
Types of Mutation
- β⁰ = complete absence of beta chain synthesis
- β⁺ = reduced beta chain synthesis
Types of Beta Thalassemia
Beta Thalassemia Minor
- Heterozygous condition
- One normal beta gene + one defective beta gene
Features
- Mild anemia
- Often asymptomatic
- Microcytosis present
Hemoglobin Pattern
- HbA₂ increased
- HbF mildly increased
Beta Thalassemia Major

- Homozygous condition
- Both beta genes defective
Pathogenesis
- Beta chain production absent or severely reduced
- Excess alpha chains precipitate in erythroblasts
Result
- Severe ineffective erythropoiesis
- Massive hemolysis
Clinical Features
- Severe anemia develops after 6 months of age
- Pallor
- Jaundice
- Splenomegaly
- Hepatomegaly
- Growth retardation
Bone Changes
- Bone marrow expansion causes:
- Frontal bossing
- Maxillary prominence
- Chipmunk facies
Blood Findings
- Severe microcytic hypochromic anemia
- Target cells
- Nucleated RBCs
Hemoglobin Pattern
- HbA absent or greatly reduced
- HbF markedly increased
- HbA₂ increased
Beta Thalassemia Intermedia
- Severity lies between minor and major.
Features
- Moderate anemia
- Occasional transfusion requirement
Pathophysiology of Thalassemia

- Excess unmatched chains damage RBC precursors.
- Bone marrow destruction increases.
Result
- Ineffective erythropoiesis
- Peripheral hemolysis
Iron Overload
- Repeated transfusion and increased absorption cause iron overload.
Complications
- Cardiac failure
- Liver damage
- Endocrine dysfunction
Laboratory Diagnosis
- Microcytic hypochromic cells
- Target cells
- Anisopoikilocytosis
Hemoglobin electrophoresis
- HbA₂ increased
- HbF increased
Genetic analysis
- Confirms diagnosis
Treatment
- Regular blood transfusion
- Iron chelation therapy
- Folic acid supplementation
- Bone marrow transplantation in selected cases
| Type of Thalassemia | Basic Defect | Genetic Basis | Main Hemoglobin Formed | Clinical Severity | Important Features |
|---|---|---|---|---|---|
| Alpha Thalassemia (1 gene deletion) | Slight decrease in alpha chain synthesis | One alpha gene deleted | Nearly normal Hb | Silent carrier | Usually asymptomatic |
| Alpha Thalassemia (2 gene deletion) | Reduced alpha chain synthesis | Two alpha genes deleted | Mild reduction in HbA | Mild | Mild microcytic anemia |
| Alpha Thalassemia (3 gene deletion) | Marked reduction in alpha chain synthesis | Three alpha genes deleted | HbH formed | Moderate to severe | Hemolytic anemia, splenomegaly |
Myoglobin
- Myoglobin is a heme-containing oxygen-binding protein present mainly in muscle tissue.
- It is found in high concentration in skeletal muscle and cardiac muscle, where it acts as an intracellular oxygen reservoir.
- Its main function is to store oxygen and facilitate oxygen diffusion within muscle cells during muscular activity.
- Myoglobin is especially important in tissues with high oxygen demand, such as the heart.
Structure of Myoglobin
- Myoglobin is a simple conjugated protein.
- It contains:
- One globin polypeptide chain
- One heme group
Globin Part
- Myoglobin consists of a single polypeptide chain containing 153 amino acids.
- This chain is folded into compact tertiary structure.
Heme Part
- One heme group is present in a hydrophobic pocket of globin.
- Heme contains ferrous iron (Fe²⁺).
Oxygen Binding
Mb+O2⇌MbO2
- One myoglobin molecule binds only one oxygen molecule because it contains only one heme group.
Comparison with Hemoglobin
| Feature | Myoglobin | Hemoglobin |
|---|---|---|
| Number of polypeptide chains | One | Four |
| Number of heme groups | One | Four |
| Oxygen molecules bound | One | Four |
| Location | Muscle | RBC |
| Main function | Oxygen storage | Oxygen transport |
Reason
- Myoglobin does not show cooperative binding.
- Oxygen binding occurs independently.
Oxygen Dissociation Curve
- The oxygen dissociation curve of myoglobin is hyperbolic.
Functional Significance
- Myoglobin accepts oxygen from hemoglobin.
- Releases oxygen only when oxygen tension becomes very low.
Physiological Role of Myoglobin
1. Oxygen Storage
- Stores oxygen inside muscle cells.
2. Facilitates Oxygen Diffusion
- Helps oxygen move from capillaries to mitochondria.
3. Provides Oxygen During Muscle Contraction
- During intense exercise, oxygen demand increases.
- Myoglobin releases stored oxygen.
4. Important in Cardiac Muscle
- Cardiac muscle depends greatly on myoglobin because of continuous oxygen need.
Clinical Importance of Myoglobin
- Myoglobin is released into blood after muscle injury.
Increased In
- Myocardial infarction
- Skeletal muscle injury
- Rhabdomyolysis
Laboratory Importance
- Early marker of myocardial infarction because it rises rapidly.
Limitation
- Not specific for cardiac muscle because skeletal muscle injury also increases myoglobin.
Difference Between Myoglobin and Hemoglobin Oxygen Curves

- Myoglobin curve is hyperbolic
- Hemoglobin curve is sigmoid
Reason
- Hemoglobin shows cooperativity
- Myoglobin does not show cooperativity